
A Practical Guide To Navigate The EU's Revised GMP Annex 1
By Ryan Murray, M.Sc., and Amanda McFarland, M.Sc., ValSource, Inc.

Over the last several years, various revisions of the EU’s Annex 11 have been released and reviewed by countless numbers of subject matter experts across our industry. The final version is now published, and it is time to take action.
Navigating the changes to Annex 1 can feel more like an exploration of the Johari window. The Johari window2 sheds light on the concept of known knowns, known unknowns, and unknown unknowns and has been adopted by many industries to identify blind spots in what we know and do not know when analyzing data. If the overarching intent of Annex 1 is to bring safe and effective medicines to the world, understanding how to align with the annex and developing a framework to minimize uncertainty will shorten our timeframe to compliance. Within the framework of the Johari window, there are four categories of information:
- known knowns: those risks that your organization is aware of
- known unknowns: risks that you are aware of but that are not fully understood and can be investigated
- unknown knowns: those risks that you have not figured out yet
- unknown unknowns: those risks you do not know you do not know.
Figure 13 illustrates the relationship between the types of information that will be encountered and how that knowledge can drive us toward compliance.
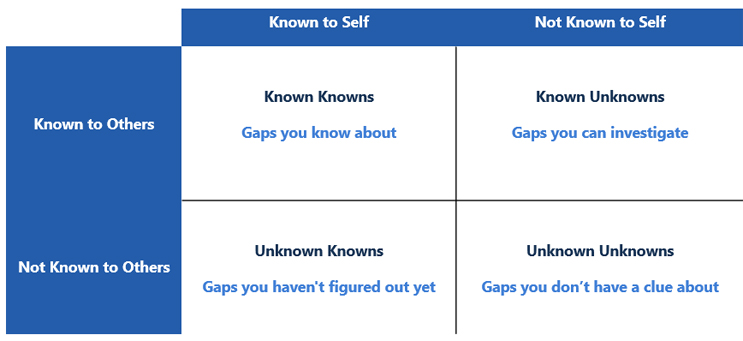
Figure 1: Johari Window Adapted for the Annex 1 Gap Assessment. Adapted from Risk Intelligence: How to Live with Uncertainty, D. Evans, 2013
This Annex 1 roadmap will use the Johari Window as a guide for uncovering information to demonstrate the increase in knowledge and understanding of your products while solving compliance challenges. Figure 2 outlines the road map basics.
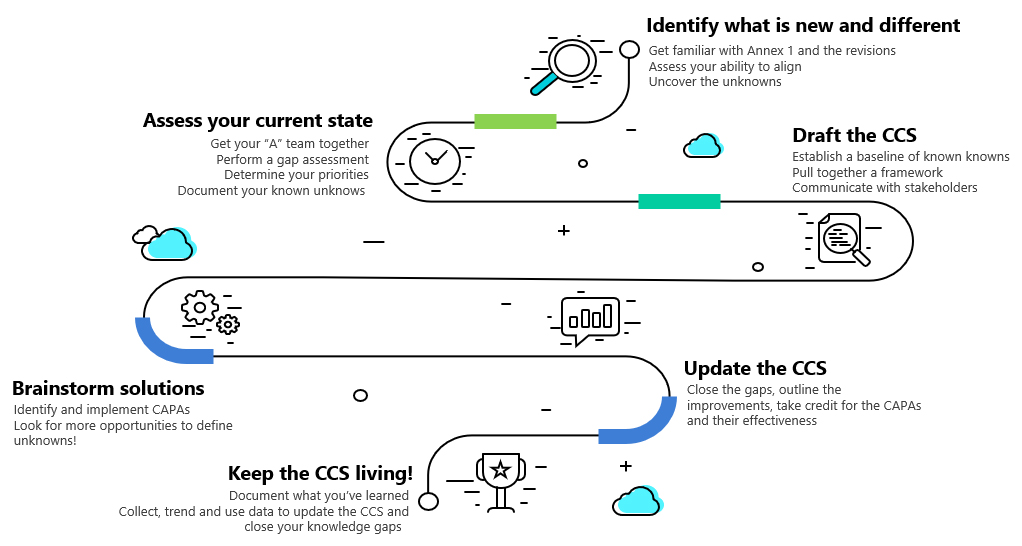
Figure 2: Annex 1 Roadmap. Click on image to enlarge.
Identify What Is New And Different: Uncover The Unknowns
Step 1: Get To Know Annex 1
The first step in developing a solid strategy for evaluating the impact of the Annex 1 revisions on your organization is to ensure that the changes to the annex are well understood and to shine a light on the unknowns. One of the key differences that exist within the Annex 1 revision is relative to the inclusion of Quality Risk Management (QRM). In previous versions of Annex 1, the references to quality risk management were limited. Now the need for risk management and risk principles are seen as foundational and fully embedded. QRM principles provide a proactive means of identifying, scientifically evaluating, and controlling potential risks to product quality. The execution of risk management and the application of risk principles comes in a variety of applications, including science and evidence-based risk assessments and multidimensional approaches to decision-making, as well as the acknowledgement and exploration of uncertainty. The necessity for QRM is most notable in relation to the contamination control strategy (CCS). The contamination control strategy is intended to be a singular document that serves as a point of reference for all the critical control points and demonstrates how a state of control is scientifically established, maintained, and remains effective. The CCS is intended to tie back to the bodies of knowledge contained within the risk assessments that helped inform the process controls to protect product quality and patient safety.
Step 2: Review Your Quality Management System (QMS)
With a firm understanding of the changes to Annex 1 and the applicability to your processes, it is time to review the quality management system to ensure it has the appropriate level of maturity to support the Annex 1 updates. Since there is a significant reliance upon QRM and knowledge management (KM) to inform the contamination control strategy, it is important to understand the current maturity of the quality risk management program. While the use of risk management has been well established through guidance documents such as ISO 310004 and ICH Q9,5 the level of maturity relative to risk management varies across the industry. Because of the intensity of risk management and knowledge management activities needed to inform the CCS, each organization needs to understand the framework by which risk assessments will be established and maintained. Inclusive of the robustness needed to support the QRM and KM programs are the frameworks related to personnel and training. Ensuring that the program has the appropriate level of management support and personnel to lead the risk assessments is only one important factor. Additional considerations include determining who will own the risk assessments and who will be accountable for mitigation and keep the risk assessments updated and reflective of the current state.
Inclusive of QMS maturity is the element of training at the site. The risk management activities needed to inform the CCS will require the site to have a group of individuals with in-depth knowledge of risk principles, experience with a variety of risk management tools, and the skills needed to lead complex risk activities.
Assess The Current State: Document The Known Unknowns
Step 3: Establish A CCS Lead And/Or Team
Navigating the creation of the CCS will require input from many facets of the organization and is not limited to the site microbiology group. Rather, to ensure a successful collaboration, a CCS lead should be identified and participation in the assessments should be multidisciplinary and inclusive of site management. The CCS lead will be the central conduit between each of the functional areas providing inputs to the contamination control strategy and coordinate the effort.
The nature of the CCS is complex and far-reaching. However, with careful coordination, the usefulness of the document will enable the organization to provide a well-constructed narrative of the state of control. There are several foundational risk assessments that will be included in the CCS related to process, environmental and critical utilities risk assessments, and aseptic process control risk assessments. The risk strategy employed, and the scope of these risk assessments must be thoughtfully considered to ensure they minimize content overlap. The team must be disciplined in both adhering to the scope defined and ensuring that the risk question defined is fully addressed. Staying on task with respect to scope and strategy will result in several efficiencies, including fewer risk assessments to update/review and maintain, minimizing the places where data are recorded/repeated to ensure consistency, maximizing subject matter expertise contribution, and reducing the burden on the document management system.
Step 4: Conduct Annex 1 Gap Assessment
Defining the differences between the previous and newly revised versions of Annex 1 will provide a structured approach to gap identification and analysis. A gap analysis tool should explore the annex requirements both at the microscopic level (i.e., line by line) and macroscopic (i.e., big picture). A line-by-line assessment will ensure that changes in language are well understood, and that the interpretation of the requirements is aligned, while the macroscopic assessment will provide a holistic view of the requirements. A holistic understanding will enable the organization to reflect on how each element of Annex 1 comes together to illustrate the full lifecycle of an aseptically produced product.
Step 5: Determine Prioritization Scheme For Gap Remediation
To ensure that the gaps identified are appropriately characterized, consider taking a risk-based approach to determining the gravity of the gap. Domains of risk to consider include product quality, patient safety, and compliance as a function of the relative risk of the gap identified. Considering these dimensions of impact will enable the organization to define the prioritization and gap remediation plan. When developing the prioritized list, consider both short-term and long-term remediation efforts. For example, if a facility gap is identified that will be mitigated in two years using automation, it is important to consider how that gap will be addressed in the interim.
Step 6: Create A CCS And Communicate Results Of Gap Assessment With Stakeholders
Once gaps have been identified and fully understood, the results of the gap assessment should be shared with all stakeholders, including site management. The identified gaps should be investigated further and a plan should be established to close the gaps. However, the CCS lead can now begin outlining the scope of the CCS document. One point to remember when writing the document is that the CCS document should serve as a roadmap for regulators, auditors, and internal stakeholders. Therefore, the CCS should not be a duplication of information already contained in another area of the quality system. Rather, a summary of a CCS element with references to other documents for much of the detail is encouraged. The CCS should include, but is not limited to, the plant and process design, equipment, personnel, utilities, raw material controls, product container closure, vendor and outsourced activity management, process and sterilization validation, cleaning and disinfection, monitoring systems, CAPA (corrective action and preventive actions), and continuous improvement practices. Each of these CCS elements should describe what controls and practices are in place and the rational for each control established.
Brainstorm Solutions: Investigate And Close The Knowledge Gaps
Step 7: Establish CAPAs
A robust CAPA6 program is a requirement for all pharmaceutical operations, let alone for Annex 1. For each gap identified, a CAPA should be established to ensure the identified gap is fully closed to meet the intent of the requirement in the annex. The team must consider the risks, not only the benefits, associated with making changes to operational strategy and/or facility/engineering controls. The team should be mindful of short-term fixes versus long-term solutions and be clear about the intent of a CAPA if it is a short-term approach. This is particularly important in older facilities where a large-scale capital improvement may be necessary to meet the expectations of the annex.
Step 8: Implement CAPAs
While creating and documenting an action plan for identified gaps may appease an auditor in the short term, this is only the first step in remediation. For a change to be impactful, it must be implemented. The timely completion of qualification or validation activities, creation of new SOPs, and training of plant operators are critical to ensuring compliance with the annex. Firms will need to fully assess the capacity of their internal resources to implement CAPAs and should consider outsourced support if needed.
Step 9: Measure Effectiveness Of CAPAs; Look For Unintended Consequences
After implementing a new change, particularly one intended to comply with a new regulatory requirement, the effectiveness of the change must be measured against the intended outcome. Unintended consequences are often a side effect of a notable change. The team should be mindful of this and actively seek this information to drive continuous improvement in the CCS, as well as the organization’s operational efficiency.
Step 10: Keep The CCS Living!
Collect, trend, and use monitoring data to maintain and/or modify the CCS in a lifecycle approach.
Data is only useful when it is analyzed. Monitoring data collected will be one of the most important tools in assessing the effectiveness of the site’s CCS. Surveillance of critical control points in the contamination control strategy should be trended to inform the maintenance of the CCS itself. A core element of the CCS is continuous improvement based on monitoring the outputs of the control strategy elements. The CCS is a living document that should be routinely assessed for its design, controls, and effectiveness. Not only will this practice identify areas to enhance product safety and quality, but regulators will also expect that periodic review of the CCS occur and the supporting data demonstrating its adequacy are available and actioned against where warranted.
Conclusion
The revisions to Annex 1 reveal a reenergized focus on risk management and proactive reflection of contamination control. The journey to compliance should include a strong strategic plan that invokes the quality system as an enabler and risk-based decision-making to prioritize the knowledge collected. The goal is to open Johari’s window to ensure that the number of unknown/unknowns are minimized while the known/knowns increase over time.
References
- EudraLex Volume 4: EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use. Annex 1 Manufacture of Sterile Medicinal Products, European Commission, Brussels, Belgium, 2022: https://health.ec.europa.eu/system/files/2022-08/20220825_gmp-an1_en_0.pdf.
- The Johari Window Model, https://www.communicationtheory.org/the-johari-window-model/
- Adapted from Risk Intelligence: How to Live with Uncertainty, D. Evans, 2013
- ISO 31000 Risk Management- guidelines, International Organization for Standardization, 2018
- ICH Quality Guideline Q9: Quality Risk Management; International Conference on Harmonization: 2006. www.ich.org
- Root Cause Investigations for CAPA: Clear and Simple, James Vesper, DHI Publishing, 2020
About The Authors:
 Ryan Murray is a quality and manufacturing science consultant with ValSource, Inc. His focus is in the areas of facility design and control, technology transfer, process qualification, and aseptic risk management in biologics and Advanced Therapy Medicinal Products (ATMPs). He is an active member of the International Society for Pharmaceutical Engineering (ISPE) and the Parenteral Drug Association (PDA) and holds a B.S. in biomedical science and an M.S. in biochemistry and biophysics degrees from Texas A&M University. He can be reached at rmurray@valsource.com.
Ryan Murray is a quality and manufacturing science consultant with ValSource, Inc. His focus is in the areas of facility design and control, technology transfer, process qualification, and aseptic risk management in biologics and Advanced Therapy Medicinal Products (ATMPs). He is an active member of the International Society for Pharmaceutical Engineering (ISPE) and the Parenteral Drug Association (PDA) and holds a B.S. in biomedical science and an M.S. in biochemistry and biophysics degrees from Texas A&M University. He can be reached at rmurray@valsource.com.
 Amanda McFarland is a quality risk management and microbiology senior consultant with ValSource, Inc. She specializes in the creation and implementation of risk management programs and developing risk-based strategies for use in clinical and commercial settings. McFarland is an active member of the Parenteral Drug Association (PDA), co-chair of the PDA ANSI standard on QRM in aseptic processing, a member of the PDA Regulatory Affairs and Quality Advisory Board, and an instructor for the PDA courses on quality risk management implementation. She has a B.S. in entomology and an M.S. in mycology, both from the University of Florida. She can be contacted at amcfarland@valsource.com.
Amanda McFarland is a quality risk management and microbiology senior consultant with ValSource, Inc. She specializes in the creation and implementation of risk management programs and developing risk-based strategies for use in clinical and commercial settings. McFarland is an active member of the Parenteral Drug Association (PDA), co-chair of the PDA ANSI standard on QRM in aseptic processing, a member of the PDA Regulatory Affairs and Quality Advisory Board, and an instructor for the PDA courses on quality risk management implementation. She has a B.S. in entomology and an M.S. in mycology, both from the University of Florida. She can be contacted at amcfarland@valsource.com.