
Bacterial Endotoxin Testing, Part 3: Calculating Endotoxin Limits & MVD
By Yadnyesh Patel, microbiology subject matter expert

With this article, I continue my article series on bacterial endotoxin testing (BET). In my first article, I provided an overview of BET, including an introduction to the Limulus amebocyte lysate (LAL) method. In my second article, I discussed the preparatory requirements for the LAL test, reagents calibration/qualification, etc. In this article, I discuss depyrogenation and calculating endotoxin limits and maximum valid dilution (MVD), including those for active substances and excipients, drug–device combination products, and medical devices.
What Is Depyrogenation?
Depyrogenation refers to the removal of pyrogens from any object. The most prevalent and problematic pyrogens are the bacterial endotoxins. Thus, depyrogenation is a process that will either destroy or remove bacterial endotoxins. Standard depyrogenation methods are dry heat, rinsing, and filtration.
Depyrogenation by dry heat is a commonly used process for depyrogenation of glassware used for BET analysis. Dry heat sterilizers are qualified with validated load patterns and used for depyrogenation of glassware in pharmaceutical industries. This is the process of destroying endotoxins through exposure to high temperatures. The heat-resistant glassware, which can withstand temperatures of 250°C or above for 1 hour, is depyrogenated and used for BET analysis.
BET results are considered valid upon the satisfactory results of all controls and tests (i.e., negative control, negative product control, positive control, and positive product control). Hence, to evaluate the BET of samples, it is mandatory to get the appropriate results of controls (NC and PC). And to get appropriate results of controls, the glassware should be depyrogenated.
Calculating Endotoxin Limits
An endotoxin limit specification for a compendial article is the allowable amount of endotoxin activity that can be safely contained in a parenteral product, according to current understanding and experimental evidence. The endotoxin limit calculation for any drug product is dependent on three variables:
- the route of administration, which largely defines K, the numerator in the endotoxin limit formula,
- the dose of the product per kilogram of body weight, and
- the duration (time) of administration. This information can be found in the package insert for an approved drug product or can be obtained from the product development group for a product that is either still in development or in early clinical trials.
An endotoxin limit specification is calculated for each drug product formulation and set of administration conditions as follows:
= K/M
K = Threshold pyrogenic dose of 5 EU/kg for most routes of administration or 0.2 EU (endotoxin unit)/kg for intrathecally administered drugs.
M = Maximum recommended dose of product per kilogram of body weight of the patient. This dose relates to the concentration of active ingredient (potency of the active ingredient) in the finished product formulation. If a product is infused or injected into a patient at frequent intervals over an extended time, then M is based on the maximum total dose administered in a 1-hour period. If the pediatric dose per kilogram per hour is higher than the adult dose, the pediatric dose must be used for the calculation.
When calculating the endotoxin limit specification, body weight is defined according to the intended patient population, which can differ in terms of geographical regions or patient populations. For example, the average adult in the United States is assumed to weigh 70 kg, whereas the average adult in Japan is assumed to weigh 60 kg. Pediatric patients could be 30 kg or below. The average weights for children can be found on the Centers for Disease Control and Prevention’s clinical growth charts. The body weight factor selected for pediatric and other special category patients should consider the worst case, i.e., the lowest body weight in targeted patient populations that can receive the greatest recommended dose. There is also a special consideration with respect to body weights for veterinary products. Veterinary drug products may be administered to a variety of different species or subspecies. Generally, the smallest animal will have the greatest dose per kilogram. Reference to the product package insert is highly recommended when establishing veterinary endotoxin limit specifications.
Different routes of administration or types of product, e.g., radiopharmaceuticals or oncology products administered per square meter of body surface, have defined values for K in the endotoxin limit calculation as described in USP <85> and above. Where the product package insert describes multiple patient populations, indications, and routes of administration, it is suggested that the laboratory calculates the limit specification for each administration and chooses the most conservative one as the endotoxin limit specification for the product.
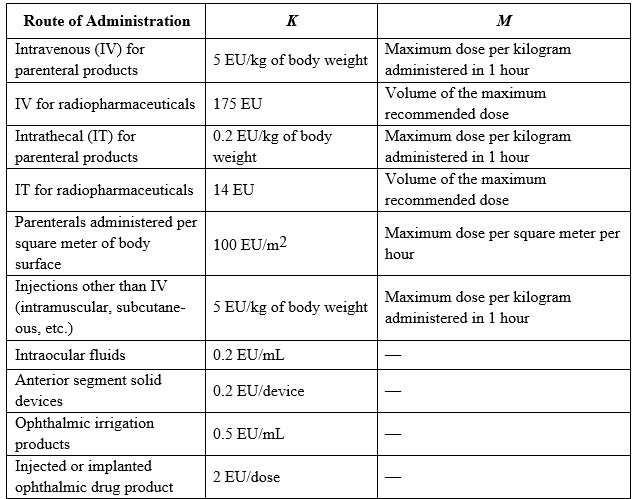
Table 1: Defined Values for K in Terms of Route of Administration.
Endotoxin limit specifications should be calculated for all indications in the product’s package insert. If a firm’s most stringent limit is lower than the USP limit, the firm should use its lower calculated endotoxin limit. It is important that the endotoxin limit specification, as a critical quality attribute for a new product, be calculated early in development and monitored throughout development and early-stage clinical trials. If a dose has not been established, the limit should be calculated based on the worst case (highest) dose that is anticipated for the product with respect to the target patient population and the route of administration. Early-phase endotoxin limits may change based on dosing and/or formulation changes prior to commercialization.
Relevance Of Limits For Compounded Sterile Preparations
Bacterial endotoxin limits can be exceeded if care is not taken when compounding pharmacies prepare injections or infusions. Additional quality control steps must be taken to avoid the addition of endotoxins to preparations made by compounding pharmacies. For example, the compounding pharmacy should only use materials with pyrogens removed in-house or they have received as sterile and free of detectable endotoxins. Only diluents from commercial vendors that meet the compendial limits for endotoxin (most often 0.5 EU/mL) should be used. The compounding pharmacy must make intrathecal (IT) preparations following strict IT endotoxin limits. Endotoxin limits are more stringent for IT formulations because the cerebrospinal fluid will be exposed to the IT preparation following injection.
Calculating Endotoxin Limits For Active Substances And Excipients
Controlling the levels of endotoxins in excipients and active substances can minimize the risk of finished drug product contamination. Suppliers and drug manufacturers should perform risk assessments of these substances based on raw material origins, production methods, representative sampling and testing, and storage conditions. For example, materials of natural origin such as sugars, heparins, and enzymes may contain significant levels of endotoxins and/or glucans. Suppliers of these materials should be audited or closely evaluated to ensure control of bioburden and endotoxins in their manufacturing operations. If these suppliers provide a COA regarding the endotoxins or glucan content for individual lots of material, the evaluation should also include an assessment of the validity of test methods, the accuracy of test results, and any testing history. Consideration should be given to using a glucan blocker for products or materials that may contain glucans in addition to endotoxins. Glucan blockers are used to ensure that all tests are specific to endotoxins, preventing a false-positive reaction of the test to glucans. Because products do not have glucan specifications, glucan-blocking reagents are specific to assay methods and lysate formulations and are generally offered by lysate reagent manufacturers. Because glucan-blocking reagents are used in different ways, it is recommended that laboratories follow the reagent manufacturer’s instructions for the use of these reagents. If non compendial materials or articles are tested and released in-house, endotoxin limits should be assigned after a thorough understanding of their potential contribution to the formulation. Working backward from the calculated drug product specification for endotoxins, limits can be assigned to each individual component in the formulation with the assurance that the drug product limit would not be exceeded if each component were at its limit. If the same component is used in multiple formulations, the lowest limit should be used for the testing and release of that component.
Calculating Endotoxin Limits For Combination Products
A combination product is defined as a product composed of two or more regulated components (i.e., drug/device, biologic/device, drug/biologic, or drug/device/biologic) that are physically, chemically, or otherwise combined or mixed and produced as a single entity. It also is defined as two or more separate products packaged together in a single package or as a unit and composed of drug and device products, device and biological products, or biological and drug products. Typically, these products can be two or more regulated components presented as a single-entity product (e.g., prefilled syringe) or a kit where two or more regulated products are packaged for use together as a single-entity product (e.g., a lyophilized drug packaged with a diluent and a syringe). Although a manufacturer can propose and justify unique limits for review in regulatory submissions, there are some general points to consider:
- If the combination product is a drug/device combination such as a prefilled syringe, the prefilled syringe may be tested as a filled unit and the endotoxin limit for the drug product prevails. Any endotoxins contributed by the container (device) are assumed to be eluted with the drug product during sample preparation and, therefore, are accounted for in the product’s assayable endotoxins.
- If the combination product is two drugs to be administered simultaneously (via IV or intramuscular [IM]), then the endotoxin content of the combined dose may not exceed the endotoxin limit for drugs of 5 EU/kg/h for IV or IM administrations or 0.2 EU/kg/h for IT administration.
- If the combination product is a kit containing multiple components that are administered as a single entity (e.g., a lyophilized product/diluent/syringe), the endotoxin content of the combined dose may not exceed the endotoxin limit for drugs of 5 EU/kg/h for IV or IM administrations or 0.2 EU/kg/h for IT administration
Calculating Endotoxin Limits For Medical Devices
In USP chapter <161>, the endotoxin limit for medical devices is assigned as 20 EU/device, except for those medical devices that come in contact with the cerebrospinal fluid, which have an assigned limit of 2.15 EU/device. Devices that contact the anterior segment of the eye should not exceed a limit of 0.2 EU/mL or 0.2 EU/device, as appropriate. Endotoxins in or on solid matrix medical devices are not measured directly, but rather the device is rinsed, soaked, or extracted in an appropriate volume of solvent (generally WFI) and the extracts are tested; in some cases, extracts from several devices are pooled for testing. As a result, the endotoxin limit for a device extract is expressed in EU/mL, which can later be converted mathematically to EU/device. The endotoxin limit for an extract is inversely proportional to the volume of solvent used for the extraction.
The relationship is:
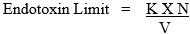
where:
K = endotoxin limit per device (20 EU unless otherwise defined and justified; 2.15 EU/device for IT devices)
N = number of devices represented in the pool
V = total volume of solvent used to extract the devices.
For example, if the laboratory tests 10 IT devices, each with an extraction volume of 50 mL, the endotoxin limit specification for the pooled extract is:
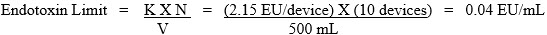
What Is MVD?
Maximum valid dilution (MVD) is the maximum allowable dilution of a sample at which the endotoxin limit can be determined.
As product-associated interferences are diluted, so will any endotoxins in the sample be diluted. Therefore, a calculation called the MVD defines the upper bound of allowable product dilution. The MVD is dependent on the endotoxin limit for the product, the starting concentration of the product (generally the concentration of the active ingredient), and the sensitivity of the test method.
The MVD is defined as:
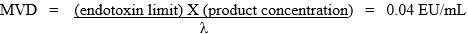
where:
endotoxin limit is the calculated limit for the product or device and
product concentration equals the concentration of the active ingredient in units per milliliter. For those products administered on a milliliter per kilogram basis or for medical device extracts, the product concentration equals 1.
λ = the confirmed label claim sensitivity for gel clot method or the lowest point on the referenced standard curve for the quantitative tests
Additional information on the MVD includes the following:
- The endotoxin limit is constant for any given formulation/dose/administration.
- The MVD is directly related to the starting concentration of active ingredient, and the higher the starting concentration, the higher the MVD.
- The MVD is inversely related to the numerical value given to the test method sensitivity. The more sensitive the test (λ as the denominator in the MVD formula gets lower), the higher the MVD. Changing the test sensitivity to a lower number (more sensitive test) will assist in providing additional dilution room for interfering products.
- The units in the formula cancel out. The resulting calculated value is a dilution factor with no units. For example, an MVD of 100 means that the product concentration can be diluted no further than 1:100 to have a valid test.
- The MVD does not limit the necessary product dilution when determining the true amount of product contamination in a sample that fails to meet the endotoxin limit. When a product fails at the MVD, it does not meet the endotoxin limit requirement. In this case, it is recommended that the laboratory determine the total bacterial endotoxin content in that product by dilution to extinction (a negative test for gel clot and a valid reading on the quantitative standard curve). The actual level of endotoxin contamination in the product may prove very helpful for trending purposes and to determine the root cause of the contamination during the OOS investigation.
These Guidelines Will Help You Throughout The BET Process:
- USP chapter <85> Bacterial Endotoxins Test
- USP chapter <1085> Guidelines On The Endotoxins Test
- ICH guideline Q4B
- General Test 4.01 Bacterial Endotoxins Test (Japanese Pharmacopoeia)
- General notices 2.6.14: Bacterial Endotoxin Test European Pharmacopoeia (Ph.Eur.)
- General notices 2.6.32: Test for bacterial endotoxins using recombinant factor C Contaminants (Ph.Eur.)
In my next article (the final article on BET of my planned article series), I’ll discuss BET analysis, including labeled lysate sensitivity, non-interfering dilution determination, validation for interfering factors, and more.
About The Author:
 Yadnyesh Patel earned his master’s degree in microbiology from KBCNM University, Maharashtra, India, in 2009. With over 13 years of extensive experience in quality functions, he has spent time working at pharmaceutical organizations such as Claris Otsuka Ltd, Sun Pharmaceutical Industries Ltd., and Zydus Life Sciences Ltd. Currently, at Zydus group, he spearheads microbiological quality functions, document management, audit, and compliance. He possesses substantial expertise in quality management systems, SOPs, documentation management, microbiological test method validations, sterility assurance, aseptic process simulation, and computer system validation.
Yadnyesh Patel earned his master’s degree in microbiology from KBCNM University, Maharashtra, India, in 2009. With over 13 years of extensive experience in quality functions, he has spent time working at pharmaceutical organizations such as Claris Otsuka Ltd, Sun Pharmaceutical Industries Ltd., and Zydus Life Sciences Ltd. Currently, at Zydus group, he spearheads microbiological quality functions, document management, audit, and compliance. He possesses substantial expertise in quality management systems, SOPs, documentation management, microbiological test method validations, sterility assurance, aseptic process simulation, and computer system validation.