
From IV To Subcutaneous: It's Not About Convenience, It's About System Design
By Monika Sharma, fractional CMC & program leader, CDO, EOSPA LLC

The shift from intravenous (IV) to subcutaneous (SC) delivery has become one of the defining development themes in biologics. The appeal is obvious. Subcutaneous administration can reduce infusion burden, move treatment closer to the home, improve patient convenience, reduce chair time and staffing pressure, and strengthen life cycle value. In several therapeutic areas, the expectation is no longer simply that a biologic can be given subcutaneously but a requirement for the product to be competitive. This is especially true where the standard of care is moving toward patient-centric delivery and where site-of-care economics are under increasing pressure.
But the teams still tend to oversimplify the transition. IV → SC is often discussed as a route-of-administration change, a formulation challenge, or a device selection exercise. Those are all real components of the problem, but none of them, on their own, are the problem. The real issue is that IV → SC is a system-design decision. It links:
|
|
Once a team starts to treat any one of those variables as independent from the others, the transition starts to drift toward friction, rework, and late-stage surprises.
That is why some programs appear technically feasible on paper and still struggle in execution. They may be able to formulate at a high concentration, identify a wearable injector capable of handling a larger volume, and generate an apparently coherent development plan. And yet, when the system is stress-tested against real-world constraints — patient tolerability, injection time, device usability, process robustness, commercial dose burden, and cost per dose — the design begins to break.
This is also why the move to SC cannot be delegated to one function. It is not just a CMC, device, or a commercial issue, it is a cross-functional decision architecture problem. Early dose assumptions made in clinical development can create downstream formulation constraints. Device assumptions made too late can force concentrations into technically unstable territory. Manufacturing decisions can protect short-term timelines while undermining long-term commercial viability. Human factors work can confirm a design that should have been challenged much earlier.
The central argument of this article is simple: the move from IV to SC should be treated as integrated system design from the beginning, not as a sequence of local optimizations. That means the right question is not, “Can we convert this IV product to SC?” The right question is, “Can we design a subcutaneous system that still works when dose, patient experience, manufacturability, regulatory expectations, and economics are all considered together?”
The Simplification Problem
In my experience, teams often start in the wrong place. The question typically asked is whether concentration can be increased enough to reduce volume, or which device can tolerate the target viscosity. Those are tactical questions. The strategic question comes earlier: what system are we trying to build, and what are the nonnegotiable constraints that system must satisfy?
That difference matters because the transition to SC is rarely limited by only one factor. High-volume SC delivery is now technically more achievable than it once was, particularly with the broader use of on-body and wearable systems that can deliver volumes in the 5 to 20 mL range over extended periods. At the same time, the rise of high-concentration monoclonal antibody products has expanded what developers view as formulation space. But capability expansion should not be mistaken for the removal of constraints. It simply changes which constraints become limiting and when.
For example, a team may conclude that a 150 or 200 mg/mL formulation is technically possible, but the fact that a formulation can be made does not mean it can be filled efficiently, remain stable through shelf life, maintain acceptable subvisible particle behavior, and still be deliverable through the intended device without an injection experience that undermines adoption. Likewise, a device may technically deliver a target volume, but if injection duration, wearability, patch adhesion, user steps, or perceived burden are not acceptable in real life, the design is not strong simply because the mechanism functions.
This is where I see many teams become overly optimistic. They assume that if each workstream can produce a technical answer, the aggregate system will work. In practice, the opposite is often true. A system can fail while every individual function can defend its own logic.

The formulation team can say the concentration is achievable.
The device team can say the injector can deliver the fluid.
Clinical can say the dose is justified.
Manufacturing can say the process is feasible.
And yet the total product may still be too expensive, too burdensome, too slow to administer, too difficult to scale, or too fragile to sustain.
That is why IV → SC is better understood as a coupled-constraints problem. It is less about any one breakthrough than about whether the full chain of decisions remains coherent under stress.
Pattern Recognition: How IV → SC Programs Start To Break
Across programs, there are recurring early signals that the transition is being handled as a component problem rather than a system problem. These signs usually appear before a formal failure point, which is why they are so important.
1. Dose is treated as fixed instead of designed.
One of the earliest and most consequential signals is when dose is carried forward from the IV paradigm without enough challenge. In IV programs, higher mg/kg dosing may be acceptable because administration time and route make larger delivered quantities tolerable. In SC development, the same assumption can become a hidden structural problem. Dose determines required delivered mass. Delivered mass, in turn, constrains concentration and/or volume. That then shapes viscosity, device selection, injection time, and cost per dose. When teams say the SC transition is difficult, they often really mean the inherited dose architecture is difficult.
2. Device thinking begins after formulation targets are already set.
Another common sign is when the concentration target is effectively set before device feasibility has been seriously evaluated. In those cases, the device is not helping define the system; it is being asked to rescue a system that has already been defined elsewhere. This often results in late-stage realization that injection time is too long, required force is too high, container format is limiting, or usability becomes compromised. At that point, options are narrower and more expensive.
3. High concentration is assumed to be a universal solution.
High concentration is often treated as the elegant answer to the volume problem. It can reduce volume, but it may also increase viscosity, aggregation risk, syringeability constraints, sensitivity to shear, and processing burden. When teams describe high concentration as the answer rather than as one element of a broader trade space, they are usually not thinking systemically enough.
4. Human factors is positioned as validation rather than design input.
When human factors enters the conversation only after a device concept is largely fixed, the program is already late. Usability should not simply validate that users can complete a task sequence. It should help shape what an acceptable task sequence is. If users are being asked to manage long wear times, multiple steps, awkward body placement, or uncertain completion cues, that is not just a human factors question. It is a system design issue.
5. Manufacturing and COGS appear in discussion only after feasibility is declared.
A final signal is when technical feasibility is declared before manufacturing reality and cost structure are seriously interrogated. Many IV → SC concepts are viable in development quantities but look very different when translated into commercial supply assumptions. High required protein mass, yields, long cycle times, specialized device assembly, and fill/finish constraints can turn an otherwise promising transition into a marginal or unattractive commercial proposition.
The System Design Framework
A useful way to frame IV → SC decision-making is to think of the transition as a linked chain of constraints:
Dose ↔ Volume ↔ Concentration ↔ Device ↔ Injection Experience ↔ Manufacturing ↔ Supply Chain ↔ COGS ↔ Commercial Viability
This chain is deliberately broader than the simpler dose-volume-concentration-device view often shown in industry discussions. It adds the elements that usually determine whether the transition remains viable once it moves beyond technical concept into a real product system. These added elements, particularly injection experience, supply chain, and economics are where seemingly coherent designs often start to unravel.
The table below shows why no single decision can be treated in isolation.
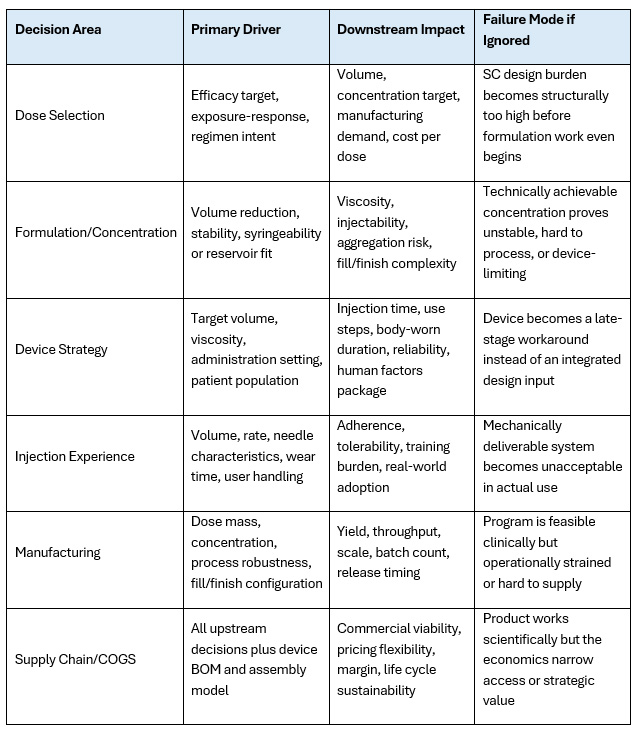
In practice, these relationships are bidirectional. Teams often speak as if dose drives the rest of the system in only one direction. In reality, the downstream design can and should push back upstream. If the intended injection experience is unacceptable at the intended dose, the response should not automatically be to search for a more heroic device or a more extreme formulation. The correct response may be to re-look at dose, regimen, target product profile, or patient segment assumptions.
In part one of this article series, we discussed the importance of considering a transition from intravenous to subcutaneous administration as a system problem rather than just a single component. In the second part of this article series, we will expand the framework and dive into what each area of a systematic program transition entails.
References
About The Author:
 Monika Sharma, M.Sc., is an executive leader with 25+ years of experience across large and emerging biotechs. Sharma brings integrated expertise across asset strategy, clinical development (early through late stage), CMC, and program execution, enabling programs to progress from IND through BLA and into commercialization. Her background spans roles from formulation scientist to vice president of product development, along with adjunct faculty positions, all of which have equipped her with the expertise to excel as an executive consultant. In her current role, as a leader at Eospa Consulting, she serves not only as an advisor but as an embedded leader, ensuring execution goes beyond planning to deliver real results.
Monika Sharma, M.Sc., is an executive leader with 25+ years of experience across large and emerging biotechs. Sharma brings integrated expertise across asset strategy, clinical development (early through late stage), CMC, and program execution, enabling programs to progress from IND through BLA and into commercialization. Her background spans roles from formulation scientist to vice president of product development, along with adjunct faculty positions, all of which have equipped her with the expertise to excel as an executive consultant. In her current role, as a leader at Eospa Consulting, she serves not only as an advisor but as an embedded leader, ensuring execution goes beyond planning to deliver real results.