
The 5 Major Regulatory Focus Areas For Medical Device Compliance In 2025
By Hilde Viroux, PA Consulting

It looks like things will be heating up in 2025 from a regulatory perspective for the medical device industry. Several rules and regulations have important due dates in 2025 and beyond for which manufacturers should start preparing.
FDA Rule On Lab Developed Tests (LDT)
The FDA rule on LDT has due dates for the coming four years, with the first one May 6, 2025.

By May 6, 2025, a laboratory that has a lab developed test (LDT) must have processes and procedures in place to collect and evaluate complaints, report MDRs, and conduct a recall. The timeline to establish all of this is very short for those who haven’t started yet. The timeline for the May 2026 due date is even shorter, as the laboratory must submit the labeling of all the LDTs as part of the establishment registration. The LDT label needs to comply with FDA’s IVD labeling requirements (21 CFR 809.10). This means that data has to be available to support the labeling claims, including, for example, specific performance characteristics such as accuracy, precision, specificity, and sensitivity. If the data is not available, laboratories should start testing sooner rather than later to have the labels ready by May 2026.
There have been industry discussions as to whether the rule will be upheld. The general consensus has been that even with a new administration in place, it is unlikely that anything will happen before May 2025. Laboratories should not count on the rule “disappearing” and must continue preparations to meet the due dates.
The FDA Adoption Of ISO 13485
The FDA issued the final rule on February 2, 2024, to amend the device current good manufacturing practice (cGMP) requirements of the Quality System (QS) regulation to harmonize with ISO 13485.
U.S.-focused companies that have only complied with 21 CFR 820 must now ensure their QMS aligns with ISO 13485 by February 2, 2026. Although it is not building from scratch, quality and regulatory personnel should be diligent in checking deviations between ISO 13485 and QMSR, paying careful attention to important distinctions in definitions. FDA announced that it will not provide a mapping of the requirement revisions between the QS regulation, ISO 13485, and the QMSR. The major differences between 21 CFR 820 and ISO 13485 are related to risk management, QMS documentation, customer satisfaction, and continuous improvement of the QMS.
February 2026 is not that far away when considering implementation of ISO 13485, even if it’s partially. A timely start will be imperative as, due to the wider scope of risk management, additional CAPAs may have to be initiated. And in the event that there is an impact on product labeling, this may be a lengthy remediation activity.
More detail can be found here.
EU MDR Extension
The original deadline for compliance with the new medical devices regulation 2017/745 (EU MDR) was May 2020 for class l non-sterile devices and May 2024 for all other devices. The dates have been postponed several times due to the pandemic, lack of accredited notified bodies, and slow uptake of the regulation.
The last extension required manufacturers to sign up with a notified body by September 2024 and then would spread out the submissions over time, levelling out the workload at the notified bodies. However, in practice, many manufacturers slowed down their compliance activities and reduced third-party compliance budgets with the intent of completing the process by leveraging their own team over a longer period. This resulted in a slowdown of the submissions of the technical documentation to the notified bodies.
Manufacturers that have strategically prioritized new product development over EU MDR compliance activities because of the extended compliance dates must be aware of the new bottleneck that this is creating for the notified bodies. The notified body review times of the technical documentation, which have already been quite long, will become even longer, extending beyond the end of the transition period. If a manufacturer submits its technical documentation one year before the due date, it most likely risks not getting its EU MDR certificates in time, resulting in potential loss of sales in the European Union.
The technical path to EU MDR from MDD is often difficult and lengthy for manufacturers, with common challenges around lifetime testing, compliance with most recent standards, clinical data, software up-classification, and cybersecurity.
The upcoming bottleneck at the notified bodies for reviewing and approving submissions is a real one. Manufacturers that plan their submissions up to 18 months before the deadline are at risk of not getting their EU MDR certificates in time to continue selling in the EU, putting patients at risk. Manufacturers that are ahead of the game will have a competitive advantage as they can continue selling and fill the gaps left by the other manufacturers. Leaders should assess their portfolios and plan ahead by securing budget and resource needs.
More detail can be found here.
EU AI Regulation
The EU AI Act (Regulation (EU) 2024/1689) is a landmark legislation that will shape the future of AI in Europe and is expected to be the baseline for similar legislation in other countries/regions. It will have a significant impact on the AI industry and society. It will set new standards and rules for the development and use of AI systems, as well as create new opportunities and challenges for innovation. The EU AI Act attempts to regulate AI in a way that balances the benefits and risks of this transformative technology. The AI act also will impact other industry sectors like the medical device industry for devices that include AI technology. Medical device manufacturers will have to comply with the AI Act, since AI that is part of a medical device will fall in the high-risk category and will require oversight by an AI designated notified body.
The EU AI Act was published July 12, 2024, and will apply by August 2, 2026. However, some elements of the act become mandatory by August 2, 2025, like the implementation of the QMS requirements, the identification of the economic operators, and the obligation to report complaints.
Medical device manufacturers that are using AI as part of their device or device software should be assessing the AI act requirements and ensuring they are built into their QMS, as the due date is less than a year out. Having a QMS compliant with ISO 13485 that satisfies medical device regulations is not sufficient. The QMS must cover cybersecurity requirements as well for high-risk AI systems to protect against tampering, data breaches, and other security risks.
Most medical devices manufacturers will have their economic operators (AR, importer, distributor) already identified and in place as this is also a requirement under the medica devices regulations. However, it will be important that contracts with those economic operators are updated to include the obligations related to the AI Act.
Full compliance with the EU AI act will be mandatory by August 2, 2026, with AI systems meeting the requirements, the sandboxes established, the notified bodies designated and fully operational.
More information can be found here.
EU IVDR
The Regulation on In Vitro Diagnostic Devices 2017/746 (IVDR) also has been amended several times, similar to EU MDR, to postpone the due dates for compliance.
However, an important date is coming up for IVD manufacturers, as full QMS compliance is due by May 26, 2025. Since most IVD manufacturers did not need a notified body under the previous legislation, they are not used to being inspected and their QMS may not be up to date. Implementing the QMS per ISO 13485 is therefore a priority to meet the due date.
The latest amendment of IVDR by Regulation 2024/1689 identifies new due dates for IVD compliance, as summarized in the table below.
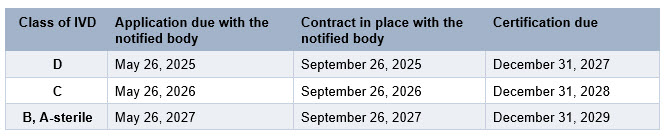
Like the EU MDR compliance journey, IVDR compliance is a lengthy and complex project. The challenges and bottlenecks identified for EU MDR apply to IVDR, with the additional challenge for IVD companies that they may not have experience with submissions and notified bodies.
To be successful requires a structured approach, as outlined in this article, which we published in 2021. Even with new compliance due dates, the activities remain quite similar. IVD manufacturers also can learn from the EU MDR implementation experience to avoid potential challenges and pitfalls. This was discussed in this article ‘3 lessons learned from EU MDR implementation to ensure IVDR adoption’.
 About The Author:
About The Author:
Hilde Viroux is a senior regulatory strategist at PA Consulting focusing on new and emerging legislation affecting medical devices and pharma companies.