
Use-Related Risk Analysis For Combination Products
By Renée Bailey, senior manager, human factors engineering, BlackHägen Design
")
In my previous article, I shared foundational concepts of implementing human factors engineering (HFE) and risk management processes with evolving regulatory expectations for combination product development. One of the most integral components discussed is the use-related risk analysis (URRA) and its applicability to the overall HFE and combination product development process. In this article, I’ll take a closer look at the URRA, the complexities it brings together, and the value it adds to product design and development.
In the simplest terms, a combination product is a medical device combined with a drug or biological product. As such, the human factors engineering (HFE) process must investigate numerous factors to ensure that the entire user interface of the combined product is safe and effective, including minimizing interactions that may result in medication errors. In its final guidance, the FDA explicitly calls out its focus on tasks that can impact dosing, administration of the product, and have the potential to result in harm. As I discussed in my earlier article, information about tasks, potential use errors, potential harms resulting from potential use errors, and the severity of those harms are all documented in the use-related risk analysis (URRA). Risk control measures are also identified, which are the design elements of the user interface intended to eliminate or reduce the occurrence of the identified risks.
To expand on this further, tasks completed by different types of users are identified in the URRA to assess risks across potential product users and their use environments. Very different risks can be associated with use errors encountered by healthcare professionals versus lay patients or caregivers. The URRA provides critical input when designing HFE evaluations so that use by all users in their representative environments and the potential risks of use errors are assessed. The iterative cycle of designing a product (even early prototypes), conducting a URRA, and evaluating the user interface through HFE is the bedrock for efficiently developing safe and effective products. The systematic approach (see Figure 1) eliminates stages of trial and error, zeroes in on specific aspects of design that are most important, and paves a smoother path to regulatory approval.
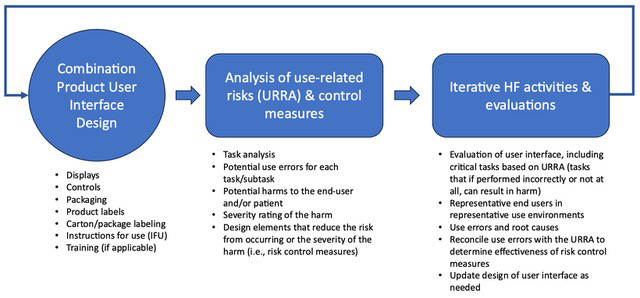
Figure 1: Iterative Optimization of Combination Product User Interface. © 2024 BlackHägen Design.
Deficiencies in the URRA are a leading cause of delays with FDA submissions, which can result in unexpected delays to market and additional costs to conduct supplemental HFE activities or evaluations. Combination products designed without the benefit of a well-crafted URRA also run the risk of not being competitive in the market and having high post-market costs to support end users and patients or to address safety concerns, recalls, etc.
Timing Is Everything: Get An Early Start With The URRA
When the FDA reviews HFE data as part of regulatory submissions, it is looking for evidence that the product has been iteratively optimized to establish that it is safe and effective in the hands of the end users. The evidence should show the risks of harm to the end user and/or patient have been designed out of the user interface as much as possible and practicable.
To produce this evidence, product teams should start early with an integrated HFE, URRA, and risk management process. Starting early ensures ample time to conduct the requisite user and environment analysis, URRA, and HFE activities, then consider design changes that are driven by the HFE data collected. This approach is ideal because design changes can be made early when the cost and effort to implement design changes are incremental compared to late-stage design changes. This early effort not only gives product teams an edge in designing a safe and effective product, but it also builds the risk management “story” needed for the HFE submission.
Unique Aspects Of The URRA For Combination Products
The complexity of combination products arises from the diversity of users, environments, delivery devices, and the combined properties of the drug and the overall user interface, as well as the potential effects of medication errors. The URRA helps product teams comprehensively examine how the drug, device, or combination of the two contribute to risks and identify opportunities to design the product to mitigate those risks. It is important to note that design opportunities exist across the user interface, including the delivery device, packaging, package labels, outer carton, instructions, training, etc. Emerging therapies for drug or biologic delivery may also include a medical procedure, such as an implant procedure. In these cases, the procedure, including any devices used for the procedure, should also be optimized for the healthcare professional(s) involved.
Below are several examples that illustrate how examining the matrix of users, environments, and attributes of the combination product using the URRA can lead to product design opportunities.
- Highly viscous drug for injection: Lay user patients could have certain physical limitations based on their age, health condition being treated, or comorbidities. For instance, users who are over 50 years of age may have age-related hand-strength limitations making it difficult for them to grasp an injector or push a viscous drug from a syringe to fill an on-body injector. These use-related difficulties should be considered during design as they could lead to the patient not administering the drug, not administering the complete dose, or a delay in their therapy.
- Dry powder inhaler: Intended users of a dry powder inhaler that uses a capsule-based powder are over 50 years of age and could experience difficulties visualizing the contents of the capsule due to age-related vision changes. This could lead to risks of not inhaling all the medicine from the capsule to get a full dose of the drug. Early evaluation of these difficulties could lead to the selection of a different dry powder capsule or changing the delivery device design to accommodate a prefilled cartridge instead of a capsule.
- Drug storage: Lay user patients are expected to store a drug product in a refrigerator. Depending on the patient’s condition being treated and comorbidities, other medications may also be stored in the same location. This leads to the risk of the patient selecting the wrong drug product, which could result in a missed dose of the intended medication and over-delivery of the other medication. The design of the packaging should be optimized to guard against spills and contamination in the refrigerator, and the package label should clearly indicate the drug product to the patient. The package label may also include information to prevent lay patients from storing the product in extreme temperatures, such as in their vehicle, where it may get very hot or very cold.
- Implantable biological product: The procedure for an implantable biological product includes several components used during the procedure and involves handling of the biological product, which can be easily damaged. If the procedure components are not used correctly, the biological product could lose its efficacy and cause delays in the procedure to retrieve more product if more is available. The risks include delays in the procedure, potentially resulting in the patient being under anesthesia for a longer period and the potential that the treatment may not be fully administered or the treatment may be ineffective. The procedure must be optimized to ensure efficiency, and the components used should be optimized to support the procedure while minimizing the potential for mishandling the delicate biological product.
The risks identified in the URRA can also be relative based on the treatment (e.g., once a month treatment versus emergency treatment), the condition being treated (e.g., mild skin condition versus fluid around the heart or anaphylaxis), or the condition of the patient (e.g., otherwise healthy patient or a patient who is compromised due to autoimmune issues or multiple comorbidities).
An injection product delivering adjunct therapy for type 2 diabetes would have a very different use-related risk profile than an implantable pain pump delivering morphine. The risks identified for the implantable pump may still be missed doses or under- or over-delivery of the drug, but the harm to the patient would be much more dire, with higher levels of severity. Similarly, an intrathecal pain pump delivering medication directly to the fluid surrounding the spinal cord compared to a device delivering medication through inhalation or subcutaneous injection could have very different overall risk profiles.
Vulnerable populations could make some tasks more critical compared to other populations of users. For example, if a patient is immunocompromised and does not wash their hands before injecting their medication, this could carry greater risks than an otherwise healthy patient not washing their hands before injecting.
The overall goal is to establish this comprehensive foundation in the early stages of the HFE and product development processes. Doing so enables product teams to conduct iterative optimization of the user interface design to minimize use issues and ensure patients receive the intended medical benefits of the medication while minimizing risks.
Accelerate Product Design To Meet Regulatory Expectations With HFE/URRA Processes
When the URRA is implemented early as an integral part of the HFE process, it lays the groundwork for a systematic, streamlined approach to iteratively improve the design of the user interface and implement risk control measures. As these measures are implemented and assessed through HF evaluations throughout product development, product teams ensure risks are reduced or eliminated to the extent possible. By applying risk-based HFE activities early in product development, the cost and effort to implement design changes are incremental compared to late-stage revisions. The integrated, iterative approach to HFE utilizing the URRA is the most effective way to reduce the potential of deficiencies in a pre-market submission to the FDA.
For product development teams, it is imperative to follow FDA guidance and embrace HFE approaches early in the design process to avoid the added costs of resolving HFE deficiencies identified by the FDA after submission. Not only are the costs of late-stage changes often staggering, but these deficiencies also impact critical time-to-market strategies when the combination product market is ripe for embracing innovative new product designs to transform healthcare procedures, with avoidable delays creating opportunities for market competition.
About The Author:

Renée Bailey is the senior manager of human factors engineering at BlackHägen Design. With nine years of experience in human factors engineering and 26 years as a Certified Instructional Technologist (CIT) and performance improvement consultant, she has worked with large-scale programs in Fortune 100/500 companies and various regulated industries, including medical device manufacturing, pharmaceutical, utilities (oil & gas, electric), finance, and food safety. Bailey has experience applying regulatory-focused human factors best practices to leading project teams, project strategies, use-related risk analysis, threshold analyses, and human factors study execution for pre- and post-market programs. Significant experience includes projects related to drug-delivery combination products.