
We're Heading Toward A Bottleneck For EU MDR Approvals; How To Get Ahead
By Hilde Viroux and Chris Illman, PA Consulting

The deadline for medical devices compliance to the new medical devices regulation 2017/745 (EU MDR) was originally May 2020 for class l non-sterile devices and May 2024 for all other devices. During those first years, the accreditation of notified bodies to EU MDR was a lengthy process, which was further slowed down by the pandemic. There was the fear that there wouldn’t be enough notified bodies to deal with all the certificate renewals. The EU Commission therefore issued the regulation 2020/561 to postpone the compliance date for class l devices to May 2021, while maintaining the May 2024 date.
However, accreditation of notified bodies continued to be a slow process. Manufacturers of medical devices were also slow in submitting their applications. This resulted in most of the technical files being submitted in 2023. Notified bodies alerted the EU Commission that it would be impossible to handle all the applications in one year with their current resources.
To avoid possible shortages of devices on the EU market, the EU Commission issued in March 2023 the regulation 2023/607 with new extended due dates per class and type of medical device. A visual representation of the timeline is presented below, via the European Commission.
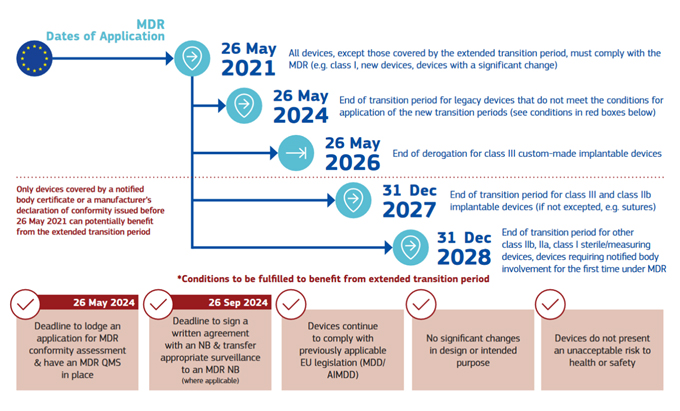
Click on image to enlarge.
The intent of the extension was that manufacturers would sign up with a notified body by September 2024 and then would spread out the submissions over time, levelling out the workload at the notified bodies.
However, what was seen in practice is that many manufacturers slowed down their compliance activities, reducing third-party compliance budgets with the intent of completing the process leveraging their own team over a longer period. This resulted in a slowdown of the submissions of the technical documentation to the notified bodies.
According to the current dashboard data from the European Commission, notified bodies signed 13,646 written agreements to extend MDD certificates, under the agreement that manufacturers would uplift those devices to MDR compliance. In April of 2022 there were 25,034 devices on MDD certificates, so over 11,000 devices were either removed from the market (over half of manufacturers report portfolio reductions because of EU MDR) or already remediated by the written agreement deadline of September 26, 2024. However, the latest tally from the 46 active notified bodies is standing at only 6,978 certificates issued out of a total 20,000+ applications, a figure that includes new devices. While this data masks the exact figures, it’s clear that the majority of MDD devices have not moved forward to MDR and have not yet made submission. The delay will result in a surge of applications that may see the review times (40% of applications now fitting in the mean time frame of 13 to 18 months) extending out significantly to two or more years and devices exiting the market as a result. This makes delaying a submission as a manufacturer very risky.
This was confirmed by Team NB, the association of medical devices notified bodies, in their press release dated October 24, 2024 where they state:
“Today, most NBs have capacity to accept applications and undertake conformity assessment activities quickly for most types of devices with some capacity restrictions for a limited number of device types. However, some NBs are facing difficulties in planning of their work and keeping their resources occupied due to delays in technical documentation submissions by manufacturers and in receiving responses to questions raised during the conformity assessment process. If such delays persist, it could lengthen the overall certification timelines and lead to potential bottlenecks in the future towards the end of the transitional timelines in 2027 and 2028.”
It is a false hope that the planned revision of the EU MDR and IVDR as called for by the EU Parliament will reduce the burden on medical device manufacturers. In the text adopted by the EU Parliament on October 23, 2024, the parliament calls for measures to make the application and approval process more efficient and transparent. However, there is no call to extend the transition period again or to reduce the amount of evidence the manufacturer must provide to ensure the safety and performance of medical devices. In more technical terms, manufacturers will still be required to fully comply with all aspects of Annex I of EU MDR, provide clinical data, and perform post-market surveillance for their devices.
Manufacturers that have strategically prioritized new product development over EU MDR compliance activities because of the extended compliance dates must be aware of the new bottleneck that this is creating for the notified bodies. The notified body review times of the technical documentation, which have already been quite long, will become even longer, extending beyond the end of the transition period. If a manufacturer submits its technical documentation one year before the due date, it most likely risks not getting its EU MDR certificates in time, resulting in potential loss of sales in the European Union.
The EU Commission is aware of this delay in submissions and the risk of shortages of certain devices on the EU market, which effectively puts patients at risk of not getting their treatment in time. It issued another amending regulation 2024/1860 in June 2024, which includes an obligation for medical device manufacturers to inform the competent authority if they anticipate an interruption or discontinuation of supply. This notification must be done at least six months in advance, and only in cases where it is reasonably foreseeable that such interruption or discontinuation could result in serious harm or a risk of serious harm to patients or public health. We expect this to result in a flurry of such notifications six months before the end of each transition period, as manufacturers will not know whether they will get their MDR certificates in time to continue supply in the EU.
The challenges facing manufacturers in the transition to EU MDR for legacy devices are manifold and evident in the application refusal rates. The first and overwhelmingly most common reason (86%) for file refusal is that under EU MDR, notified bodies may not hold accreditation for certain types of devices — this is determined on a device type basis and not by device class. This means that a manufacturer’s incumbent notified body may not have the correct designation to issue certificates it held under MDD and a new notified body must be sought — a major delay in obtaining certification.
The technical path to EU MDR from MDD is often difficult and lengthy for manufacturers, and the following are some common challenges:
- Lifetime testing — General Safety and Performance Requirements mandate safety and performance over the lifetime of the product. This makes a strong implication in many cases for life testing, which many products did not have under MDD. Whether it entails accelerated aging in single-use disposables, long-term life testing, and Weibull analysis in mechanics or HALT testing of electronics, it’s a lengthy process.
- Design change — For legacy products with a history of design changes, even the best documented history can present a protracted task around establishing quality input documents to form a coherent design history file.
- Standards compliance — Several critical standards and regulations have been updated in recent years.
- Biocompatibility, ISO10993 series, REACH, and PFAS — Continued scrutiny around materials in use can necessitate a complete product redesign and repeat verification and in some instances validation.
- Electrical and ISO60601 developments — Constant restrictions around EMC and RF in the face of ever more sensitive electromagnetic environments may go as far as to render legacy electronics obsolete. More niche stipulations of electrical standards, such as developing alarm tones, may necessitate partial redesign and impact human interaction, requiring a measure of validation update.
- Human factors and ISO 62366 — Many older products existing under User Interfaces of Unknown Provenance (UOUP) under Annex C of IEC 62366-1 may transition to EU MDR under that derogation. When the product requires substantial redesign for other compliance reasons, it can become challenging to justify grandfathering in a product or relying on historical market surveillance and summative evaluations may become necessary.
- Clinical data for legacy devices — Manufacturers often struggle to generate clinical data for devices that are on the market for a long time in a manner that will satisfy notified body expectations. Expensive post-market clinical studies may be required for the higher-risk devices.
- Software — A major shift in MDR is classification of software under Annex VIII of Regulation (EU) 2017/745, which typically results in the up-classification of software, especially where it is driving or influencing the use of a medical device.
- Cybersecurity — Where the FDA defines specific requirements, MDR operations are far more nebulous, and compliance with one does not guarantee compliance with others. Developing a robust cybersecurity case in an MDR file is risk-based (per ISO14971) and leans heavily on best-practice and state-of-the-art to achieve compliance, drawing on IEC62304 and other relevant directives (such as the Radio Equipment Directive) as guidance. Compliance by design and penetration testing can be onerous for legacy devices.
The upcoming bottleneck at the notified bodies for reviewing and approving submissions is a real one. Manufacturers that plan their submissions up to 18 months before the deadline are at risk of not getting their EU MDR certificates in time to continue selling in the EU, putting patients at risk. Manufacturers that are ahead of the game will have a competitive advantage as they can continue selling and fill the gaps left by the other manufacturers. Leaders should assess their portfolios and plan ahead by securing budget and resource needs.
 About The Authors:
About The Authors:
Hilde Viroux is a senior regulatory strategist at PA Consulting focusing on new and emerging legislation affecting the regulatory function.
 Chris Illman is a biomedical engineer based at PA Consulting’s Global Innovation and Technology Centre in Cambridge, England. He has years of experience developing new products and bringing existing products into compliance.
Chris Illman is a biomedical engineer based at PA Consulting’s Global Innovation and Technology Centre in Cambridge, England. He has years of experience developing new products and bringing existing products into compliance.